


Mutaciones del gen CFTR
En la actualidad se han identificado más de 2000 mutaciones en el gen CFTR. La deleción de la fenilalanina en la posición 508 de la glicoproteína: F508del, es la mutación más frecuente a nivel mundial; presente, en promedio, en el 70%de los alelos FQ. Un grupo de tan sólo 20 mutaciones aparece con frecuencias individuales superiores al 1% y el resto son muy raras o, en algunos casos, características de un determinado grupo poblacional.
En la Base de Datos de las Mutaciones de la Fibrosis Quística se registran las siguientes frecuencias: las mutaciones que provocan el cambio de un aminoácido por otro (missense) representan un 40.00% de las mutaciones asociadas a la enfermedad; un 8.22% las que provocan un codón de terminación prematuro (nonsense); las inserciones o deleciones que causan un cambio en el marco de lectura (frameshift) un 15,79% y aquellas que alteran nucleótidos conservados en sitios de unión intrón-exón y afectan el procesamiento del pre-ARNm (splicing) un 11.57%. Existen 2.59% de reordenamientos genómicos (grandes deleciones o inserciones), y otro 0.76% que afecta la zona promotora. Un 13,65% de cambios neutros denominados polimorfismos no ocasionan enfermedad. Las mutaciones del gen CFTR no identificadas podrían estar en intrones o en regiones regulatorias, o corresponden a reordenamientos genómicos como las grandes deleciones.
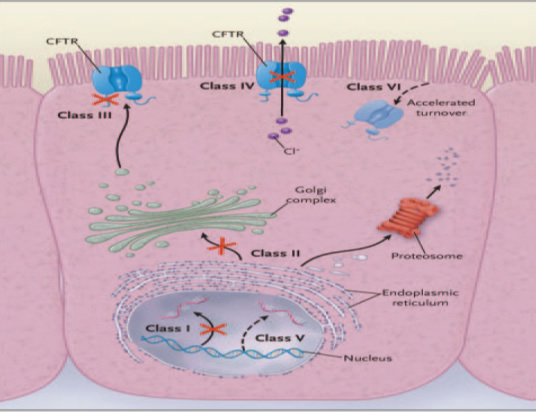
Desde el punto de vista funcional, las mutaciones en el gen CFTR pueden ser divididas en seis clases:
• Clase I: defecto en la síntesis de RNAm estable que resultan en ausencia de la proteína. (Mutaciones que producen terminación prematura de la proteína, eliminación de exones, splicing aberrante del RNAm, desplazamiento del marco de lectura). Ejemplos: G542X, W1282X, R1162X, 1717-1G®A.
• Clase II: comprende a las mutaciones que presentan un procesamiento defectuoso de la proteína, lo que impide su correcta localización en la membrana celular. Ejemplos: F508del y la N1303K.
• Clase III: mutaciones que generan una regulación defectuosa del CFTR como canal de cloruros. Ejemplo: G551D.
• Clase IV: estas mutaciones afectan la conductividad a través del canal. Muchas de estas mutaciones involucran residuos con carga eléctrica, en general, arginina, en los dominios transmembrana. Ejemplos: R117H, R334W, R347P.
• Clase V: estas mutaciones destruyen o crean sitios consenso de splicing, provocando un déficit cuantitativo del canal. Ejemplo: 3849+10Kb.C? T
• Clase VI: mutaciones que alteran la estabilidad de la proteína madura.
Aunque es imposible establecer una correlación absoluta del fenotipo con un genotipo específico, parece existir una correlación entre el porcentaje de función normal del CFTR y la manifestación de enfermedad en varios órganos. Esta información puede ser útil en la evaluación de un paciente con síntomas sugerentes de fibrosis quística pero con pruebas de sudor normal.
La heterogeneidad clínica de la fibrosis quística debe interpretarse a partir de la heterogeneidad alélica del gen CFTR, efectos de otros genes modificadores y factores no genéticos.
Los pacientes homocigotos o heterocigotos compuestos por dos mutaciones severas (Clases I, II o III), manifiestan insuficiencia pancreática, mientras se espera que un 20% de pacientes suficientes pancreático porten una mutación leve en uno de sus alelos (Clases IV o V). En la actualidad, a las presentaciones monosintomáticas, se las denomina enfermedades relacionadas al regulador de transmembrana CFTR, tales como la ausencia congénita de conductos deferentes (CBAVD), la pancreatitis crónica idiopática y las bronquiectasias, entre otras.
El número e incidencia de las mutaciones que causan FQ varía ampliamente según el origen étnico y la localización geográfica de cada población (The Cystic Fibrosis Genetic Analysis Consortium, CFGAC: http: www.genet.sickkids.on.ca/cftr).
En la Provincia de Buenos Aires, el análisis estadístico de 240 muestras estudiadas en el Laboratorio de Biología Molecular del Hospital Interzonal de Agudos Especializado en Pediatría “Sup. Sor María Ludovica” de La Plata, (Centro Provincial de Fibrosis Quística), determinó que la incidencia de las principales mutaciones analizadas fue semejante a la población del sur de Europa. El 56.0% de los alelos presentaron F508del, G542X 7,23%, R334W 2.27%, I507del 1,44, G85E 1.24%, W1282X 1.24%, 1717-1G>A 1,24, entre las más frecuentes.
Es importante destacar que, siendo la Fibrosis Quística una enfermedad autosómica recesiva, la detección de dos alelos mutados (una mutación en cada copia del gen CFTR) es diagnóstico de certeza. Sin embargo, la no detección de mutaciones no excluye la enfermedad ya que, los estudios disponibles se limitan en general al análisis de las mutaciones más frecuentes, con una sensibilidad entre el 75-80% de detección de los alelos mutados.
Un aporte importante para la interpretación de las consecuencias clínicas de una determinada mutación es el proyecto CFTR2 (Clinical and Functional Translation of CFTR, http://www.cftr2.org), el cual tiene por objeto proporcionar información clínica y funcional completa, avanzada y revisada por expertos, sobre un gran conjunto de mutaciones del gen CFTR.